High Performance Computing
This is our core area of research and expertise. High performance algorithms are necessary to allow the highly expensive calculations involved in computational chemistry to be applied to large chemical systems that are essential for the environmental effects of real systems. Much of our research has resulted in software in the widely used packages NWChem (developed primarily at Pacific Northwest National Laboratory, PNNL) and GAMESS (developed primarily at ISU in Mark Gordon’s group). A few examples of my research in this area include the development of highly scalable ab initio Monte Carlo methods that take advantage of multiple levels of parallelism, implementation of two-electron repulsion integrals onto GPU architectures, understanding how algorithm profiles effect power usage – a key issue for very large computational resources, and development of task- and data-flow execution in very expensive coupled cluster methods.
In addition to our research, Theresa has been in leadership positions related to high performance computing including co-organization of three high-performance workshops at National American Chemical Society meetings (and one that is proposed), co-chair for the DOE Exascale Requirements Review for Basic Energy Sciences, co-organizer of several workshops on hardware-software co-design, subject area editor for Parallel Computations, member of program committees for the IEEE International Parallel and Distributed Processing Symposium and the SuperComputing conference (the two leading meetings for high performance computing), member of an NSF task force on high performance computing, and member of advisory boards for several high performance computational materials and chemistry projects.
Recently, DOE has announced the Application Development projects that have been awarded for funding as part of the Exascale Computing Project to advance our software technology to the exascale (1018 floating point operations per second – believed to be the processing power of the human brain at the neural level). Theresa is part of two of these projects: “NWChemEx: Tackling Chemical, Materials and Biomolecular Challenges in the Exascale Era”, where she is the deputy director of the project, and “Enabling GAMESS for Exascale Computing in Chemistry & Materials”, where she is a part of the ISU team. In both of these projects, she will be intricately involved in and leading portions of the refactoring of the software so that the design will scale appropriately to exascale computers. In addition, she will be developing reduced cost methods and new memory models for the NWChemEx project and implementing new power aware software in the GAMESS project. These projects are exciting in the algorithmic methods to be developed and their application to catalysis science that has not been accessible with our current computational environments, both hardware and software.
top
Software and Data Interoperability
Related to the high performance computing efforts, we have been extensively involved in the development of software interfaces and data that enables multiple codes to work together. One of the largest efforts was associated with the DOE sponsored Common Component Architecture (CCA) project where we developed standard interfaces for Gaussian based integrals, energies, gradients, Hessians, optimization algorithms, multi-level parallel algorithms, density and overlap matrices, new accurate methods, combining quantum mechanics with molecular mechanics, the effective fragment potential, and database methods. In addition, we have been involved in efforts to develop informatics infrastructure for multi-scale chemical sciences, interfaces of quantum mechanical programs with dynamics programs, and a web portal for Gaussian basis sets that are essential to most of the calculations in quantum mechanics.
One of the great challenges, however, in this area of research is the adoption of interfaces and methods by others. Unfortunately, for many years the chemistry community has had only pockets of success in developing interfaces and standards that are adopted by the full community. However, within the last three to five years, there has been a shift in the community perspective. Realizing that there are immense opportunities for new scientific models and capabilities by joining forces, the community realized that it can’t keep reinventing the wheel and must work together in a synergistic manner to advance the field. Theresa is a co-Director of the newly funded NSF Molecular Sciences Software Institute (MolSSI) that will be a center for science, education, and cooperation to serve the community of computational molecular sciences. Theresa will be in charge of enabling the integration of software and data aspects for the community based on my previous research in this area. We are initially focusing on the interfaces between quantum and molecular mechanics to enable new types of simulations that are not currently possible. We are anticipating that MolSSI will be a catalyst for bringing together the molecular sciences community to establish interfaces and standards.
top
Methods for accurate energies
Extension of the CEEIS to include many-body approach
Recently, we have been working with Professor Klaus Ruedenberg to extend his Correlation Energy Extrapolation by Intrinsic Scaling (CEEIS) method to allow for excited states and to use a many-body approach to reduce the overall computational cost of large active space configuration interaction methods. Applications to date include F2 and O3.
Accurate Potential Energy Curve for C2
Diatomic carbon, C2, is found in hydrocarbon flames, comets, and the interstellar medium. C2 has an interesting electronic structure, including several low-lying excited states, and a ground state which exhibits strong multiconfigurational character even at equilibrium. The potential energy curve (PEC) of C2 dissociation is being calculated using the correlation energy extrapolation by intrinsic scaling (CEEIS) method. CEEIS was developed by Ruedenberg and Bytautas to estimate the full configuration interaction energy. The method employs a linear extrapolation between a series of configuration interaction calculations with truncated virtual orbital spaces. CEEIS has already been used to obtain high accuracy correlation energies in several diatomic systems (F2, O2, B2). Additional corrections to the PEC (core-electron correlation, scalar relativistic effects, spin-orbit coupling) were applied to these systems to obtain near spectroscopic accuracy. Highly accurate PECs for the ground state, well as several excited singlet states, were calculated as well as the vibrational frequencies for the surfaces.
top
Heavy metal systems
Critical Materials Institute
For the last two years, we have been part of the Critical Materials Institute where we have been developing software for the extraction of rare earth elements and distinguishing the binding in adjacent lanthanide complexes. One of our first studies involved examining organophosphine oxide conformations and developing molecular mechanics potentials. Using these potentials we then used computer-aided molecular design methods to design bis-phosphine oxide lanthanide extractants. Experimentalists at Oak Ridge National Laboratory are currently synthesizing the predicted extractants to determine the correctness of the predictions. In the next couple of years on the project we will be examining the effect of solvent on the preferential binding of specific metals.
Uranyl systems
here are many challenges in computationally examining heavy element systems such as appropriately describing relativistic effects, dealing with the many electrons in the system and determining an appropriate level of theory to get accurate results. Another difficulty is that there is very little experimental evidence to compare against in the gas phase. This is an important issue since it is very difficult to calibrate computations when there is limited experimental data. In a collaboration with experimentalists from Idaho National Laboratory and Lawrence Livermore National Laboratory and computational scientists from PNNL, we have examined the complexation, ligand competition and spectroscopy of uranyl, UO22+, with many different ligands including acetronitrile derivatives, acetone derivatives, and water. Our computations verify and help to explain the complexes formed in the experiments (collision-induced dissociation mass spectrometry) as well as the thermodynamic stability of nitriles relative to water. In addition, our research has helped to explain an interesting role that diacetone alcohol was playing in the experimental results where there was a question of whether there were hypercoordinated acetone uranyl complexes or not. Computations clearly showed that the uranyl acetone complex was not hypercoordinated. Our results and later experiment confirmed that the diacetone alcohol was being formed in the reactions.
top
Malonic acid tautomerism
Malonic acid is a common organic aerosol found in the atmosphere. It exists in two tautomers, a ketone and enol form. Malonic acid can react with other atmospheric substances and its reactivity may be linked to the more common tautomer in a highly concentrated deliquesced particles. Using DFT, B3LYP allows for the elucidation and confirmation of IR frequencies indicating a more abundant enol structure in the malonic acid particles. DFT is useful in this application due to the need for calculations on large systems of hundreds of atoms or more. To efficiently sample many structures on the potential energy surface (PES) and to find several low energy conformations, effective fragment potential (EFP) methods with simulated annealing in GAMESS are employed.
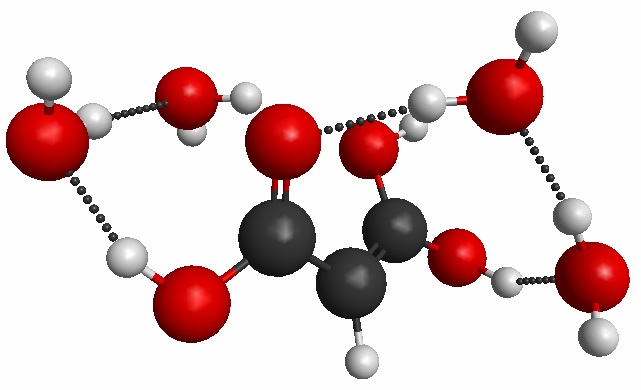
top
Chalcogen binding on coinage metal surfaces
In a collaboration with Professor Pat Thiel, Professor James Evans and Dr. Da-Jiang Liu, we are examining the effect of sulfur on the (111) and (100) faces of coinage clusters. These first two studies showed that the experimentally determined preferential binding of S to the four-fold hollow sites of Cu(100) instead of the three-fold hollow sites of Cu(111) was related to a weaker antibonding interaction in the former. In recently submitted work, we have examined small Au-X2 complexes on the Au(111) surfaces. Interestingly, the binding energies per sulfur in the gas phase are similar, but the different interactions with the surface determine whether those clusters will be stable on the surface or not. Currently, we are examining the stability of small metal-sulfur complexes both in the gas phase and adsorbed on a metal surface.
top
Cellulose
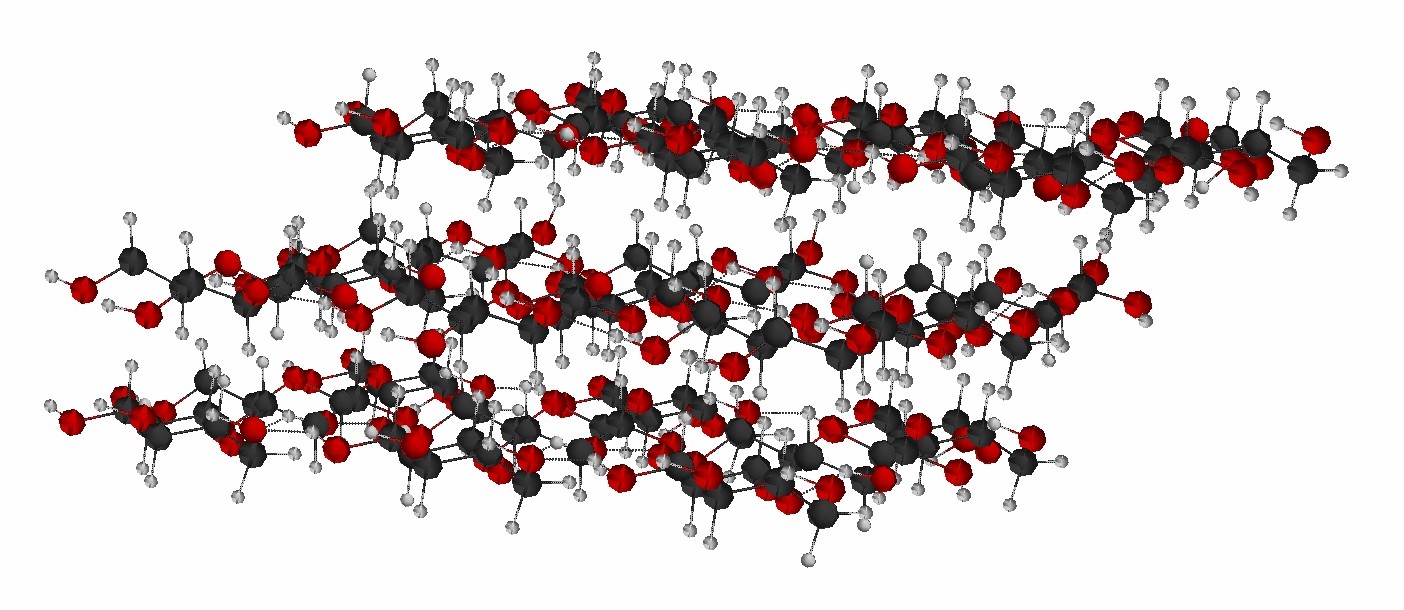
One project uses B3LYP and two-body fragment molecular orbital (FMO-2) calculations of energies to simulatethe rotation of dihedral angles formed by free hydroxyl groups in glucose residues of a cellulose fragment, and the rotation of torsion angles involving glycosidic linkages between glucose residues in cellulose (psi and phi angles). The use of FMO-2 allows us to pinpoint specific interactions among the glucose residues in cellulose that give rise to critical points in the potential energy curves obtained from the changing cellulose geometries.
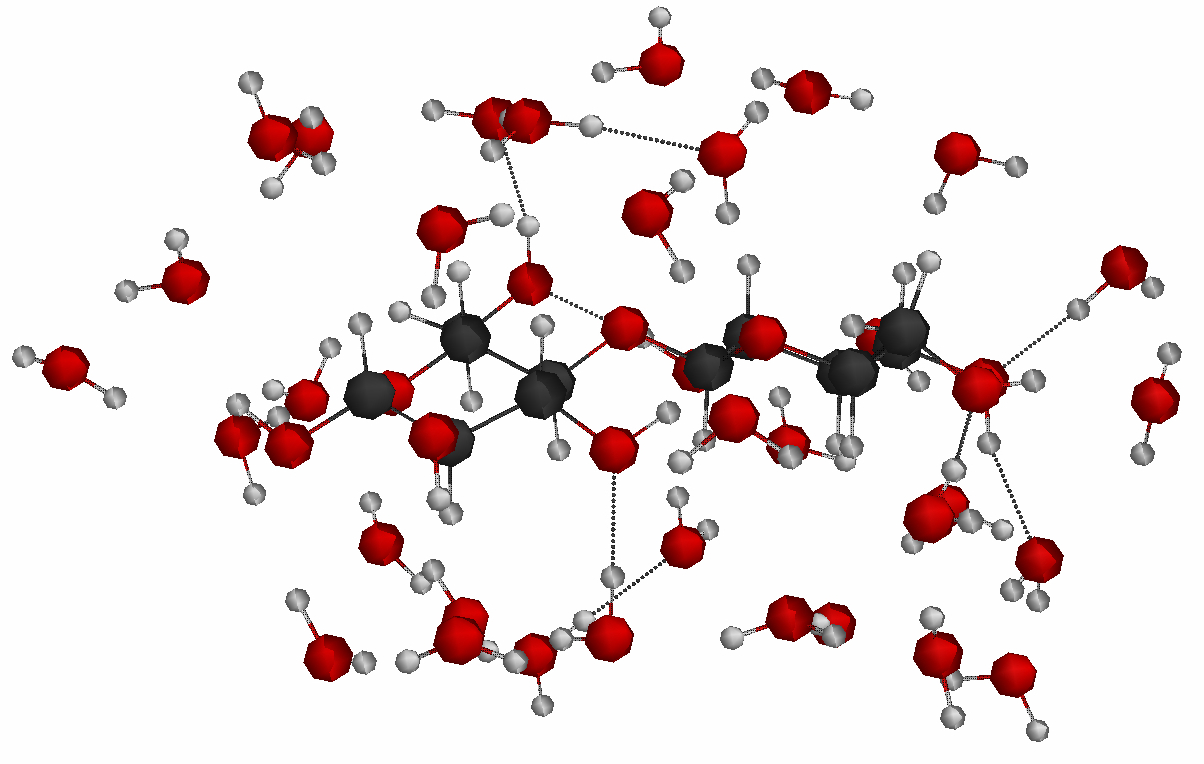
In another project, the reaction mechanism of the acid-catalyzed hydrolysis of cellobiose, the repeating unit of cellulose, is explored. In this project, EFP1 (effective fragment potential) water molecules are used to form a first solvation shell around the cellobiose molecule. The polarizable continuum model (PCM) is used to form a second solvation shell.

|
|
|
Fig A. I-alpha -12chains -144 units) FMO-study
|
Fig B. alpha-233- Cellotetraose FMO study
|

|
|
|
Fig C. celluloseIII +h2o ( 2 ns-- MD-charmm Potential)
|
Fig D. 8CelA + nanocellaose (4ns MD Charmm27 potential)
|
A. Molecular origins of cellulose recalcitrance: The goal is to understand hydrogen bonded network in polymorphs of cellulose using complex electronic structure theory methods. First FMO studies of cellulose. (Fig. A)
B. Using model of cellulose-assembly: The goal is to obtain FMO based statistical weights for strengths of hydrogen bonded interaction (case study cellotetraose)--(Fig. B)
C. All atom, FMO and CGMD simulations of crystalline cellulosic materials with explicit water: chain dynamics(Fig. C)
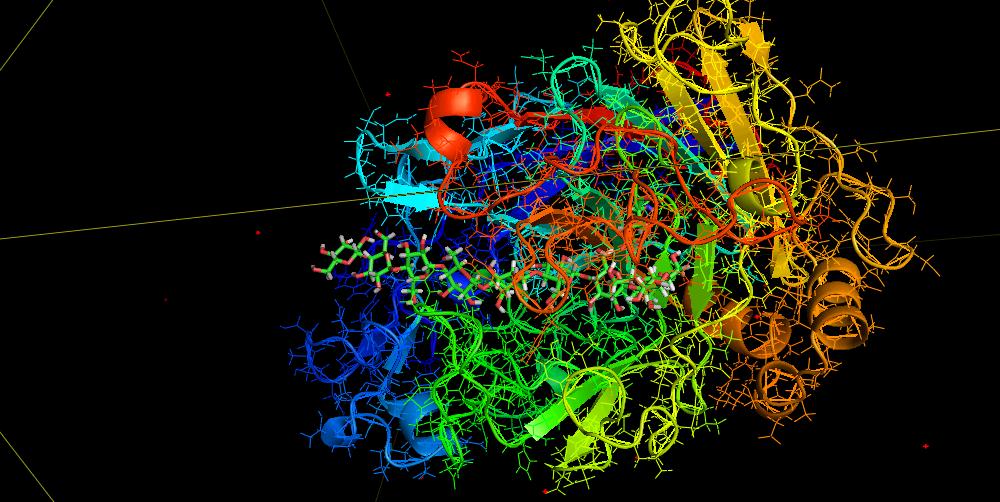
D. Biohydrolysis of cellulose: cellulose-protein binding dynamics using the all atom MD and FMO method (Fig. D)